基础函数
基础函数¶
1.输出gmt文件¶
outputGmtFile()函数中description默认为NA,如果指定,应该是一个长度与input相同,用于描述每个基因集的字符串向量。filename应该是一个.gmt结尾的文件名称,可包括路径。input是一个list或是一个data.frame,如果是list,list中每一个对象是一个向量(基因),每一个对象应该有一个合适的名称,相当于基因集的名称,下面是一个input接收list数据对象案例:
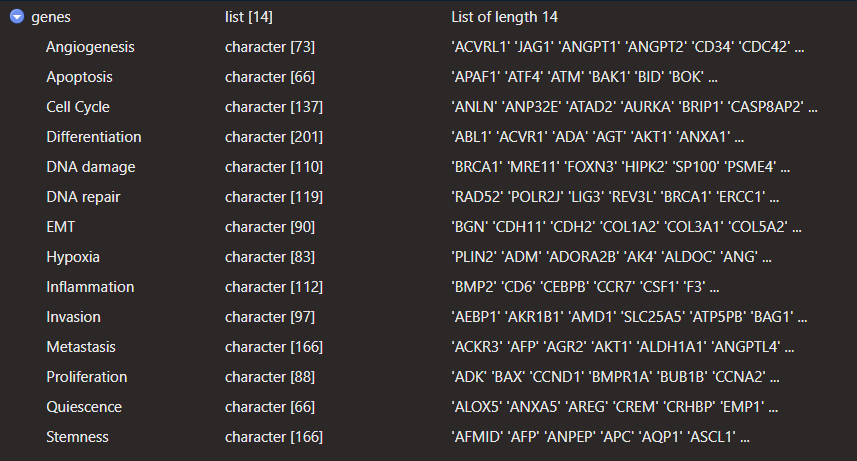
outputGmtFile(input = genes,description = NA,filename = "./gs.gmt")
如果input是一个数据框,应该包括两列,第一列是关于基因集描述的术语,第二列是基因,这时,如果需要指定description的值,长度应该等于input第一列值作为集合的长度。下面是一个input输入作为数据框的案例:
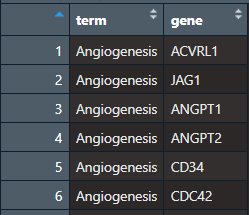
outputGmtFile(input = cg,description = NA,filename = "./gs.gmt")
2.将一个基因集生成一个gmt文件¶
geneset2gmt(geneset,genesetname,description = NA,return = "data.frame",folder= ".",filename)
geneset是一个向量,向量的每一个值应该是一个基因名称,也可以是以.txt结尾的文件路径(文件内容应该是一行一个基因名称),genesetname为这个基因集取的名字,需要指定;return的值为"data.frame"或者是"GeneSetCollection",表示什么样的值,相当于分别用clusterProfiler::read.gmt()函数和GSEABase::getGmt()函数读入gmt文件的结果,可以根据后续需要选择返回结果;该函数内部调用outputGmtFile,其他参数同outputGmtFile()。
3.整理gmt文件¶
tidy.gmt()用于统计或者合并多个gtf文件。filepath是文件路径,不用指定到具体文件,函数会自动匹配文件夹下所有以".gmt"格式结尾的文件,当然,filepath也可以是一个包括多个gmt格式文件完整路径的向量,比如:c("./gs1.gmt","/home/user/data/gs2.gmt","/home/user/geneset/gs3.gmt");fun的值是"stat" 或 "merge"中的一个,"stat"表示统计每个gmt文件中的基因个数,并输入一个csv文件,"merge"就是合并多个基因集,会返回一个数据框(不做统计,相当于函数clusterProfiler::read.gmt()的读入结果),同时输出一个gmt格式文件,无论那个,都会输出一个.unique.txt的文件以记录所有基因集中的文件;termName用于指定当fun = "stat" 时,所有基因集的统一名称,相当于列名,默认NULL(输入的列名是trem);Source是所有基因集的来源,默认"",长度应该为1;filename可以不指定文件后缀名,指定后缀名,输出也会根据fun的不同而添加相应的后缀名。addTotal的值为TRUE或FALSE,只有fun = "merge"时被使用,表示是否将所有基因集合并作为一个新的基因,相当于在输出的gmt文件中多了一行,此时作为基因集的名称由termName指定,如果termName为NULL,那么新组合的基因集名称为Total gene。
filepath <- "G:/publicData/base_files/GeneSet/Cytoskeleton/"
folder <- "G:/myProject/data/geneset"
gsdf <- tidy.gmt(filepath
,fun = "stat"
,Source = ""
,addTotal = TRUE
,termName=NULL
,save = TRUE
,folder = folder
,filename = "geneset"
)
4.整理Reactome数据框下载的通路信息文件¶
tidyGene.fromeReactome()与tidy.gmt()类似。Source默认为"Reactome";需要注意的是,下载的数据文件名以通路名称[通路ID]的形式命名,文件格式为tsv。fun的值是"stat" 时返回数据比tidy.gmt()多了PathwayID列;fun = "merge"时,2者输出一样。其他参数同tidy.gmt()。
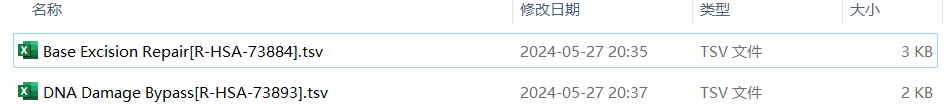
filepath <- "G:/publicData/base_files/GeneSet/DNA_Repair"
folder = "G:/myProject/DNA_Repair"
df <- tidyGene.fromeReactome(filepath = filepath
,fun = "stat"
,Source = "Reactome"
,addTotal = TRUE
,termName=NULL
,save = TRUE
,folder =folder
,filename = "geneset")
5.从基因组文件gtf中提取基因相关信息¶
gtf参数是一个gtf文件路径。
(1)提取基因长度信息¶
gtf_file <- "path/to/your/file.gtf" # 替换为实际的GTF文件路径
gene_lengths <- getGeneLenFromeGTF(gtf_file)
(2)提取基因类型信息¶
genetype <- getGeneTypeInfoFromeGTF(gtf_file)
(3)提取基因的基础信息(长度 + 类型)¶
geneinfo <- getGeneBaseInfo(gtf)
6.RNAseq数据转换¶
参考:RNAseq数据分析中count、FPKM和TPM之间的转换 (qq.com)
函数RNAseqDataTrans():
RNAseqDataTrans(data,tfun,species,gtype)
data是RNAseq数据,行可以是 'Ensemble' or 'Symbol'类型,列为样本;
tfun must be one of "Counts2TPM", "Counts2FPKM", or "FPKM2TPM";
species的值可以是 "hsa"、"mus";
gtype must be one of 'Ensemble' or 'Symbol'.
类似的函数RNAseqDataConversion
data <- RNAseqDataConversion(data,type,species = "homo",gtf = NULL)
data是RNAseq数据,行为基因(symbol),列为样本;type must be one of "Counts2TPM", "Counts2FPKM", or "FPKM2TPM".
species的值可以是 "homo"、"mus",或者是NULL。如果你的数据是人或者小鼠,只需要指定为 "homo"或"mus",如果不是,species设置为NULL,同时提供gtf格式的参考基因组文件。
7.基因集对象转换¶
read.gmt.to.getGmt()实现clusterProfiler::read.gmt()函数读入的数据转换为GeneSetCollection对象【相当于GSEABase::getGmt()读入gmt文件的结果】。genesetdf是一个数据框,有两列数据(term和gene)。
gs <- read.gmt.to.getGmt(genesetdf)
GeneSetCollection.to.df()函数相反,将GeneSetCollection对象转换为一个数据框:
gsdf <- GeneSetCollection.to.df(GeneSetCollection)