TCGA数据挖掘
TCGA数据库数据挖掘相关函数¶
TCGA数据库首页:GDC Data Portal Homepage (cancer.gov)
1.下载RNAseq数据¶
getTCGA_RNAseqData()返回一个list,包括count,tpm和fpkm 3个数据框。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD",save = TRUE,folder = ".")
2.下载蛋白组数据¶
getTCGA_ProteinExp()返回一个数据框。
Proteome_data <- getTCGA_ProteinExp("TCGA-LUAD",save = TRUE,folder = ".")
3.下载SNV(simple nucleotide variation)数据¶
数据类型为:Masked Somatic Mutation。
snv.dat <- getTCGA_SNV_Masked_data("TCGA-LUAD",save = TRUE,folder = ".")
4.下载miRNA数据¶
参考文章:
https://mp.weixin.qq.com/s/__EjCrJFc08itoF3xqawNg
https://mp.weixin.qq.com/s/-FH0Vi4PaCjhPbEq4-lxbg
https://mp.weixin.qq.com/s/WxgMhwpMAJy_CKTqNdFj0g
(1)Isoform Expression Quantification¶
IsoformEQ <- getTCGA_miRNA_IsoformEQ("TCGA-LUAD",save = TRUE,folder = ".")
(2)miRNA Expression Quantification¶
miRNAEQ <- getTCGA_miRNAEQ("TCGA-LUAD",save = TRUE,folder = ".")
5.下载甲基化数据¶
getTCGA_MethylationData 下载Methylation Beta Value数据。
MetData <- getTCGA_MethylationData("TCGA-LUAD",save = TRUE,folder = ".")
6. 下载CNV(Copy Number Variation)数据¶
getTCGA_CNV.data()函数还在优化中:
cnv.gl <- getTCGA_CNV.data("TCGA-LUAD",save = FALSE,folder = ".",data.type = "Gene Level Copy Number")
cnv.gls <- getTCGA_CNV.data("TCGA-LUAD",save = FALSE,folder = ".",data.type = "Gene Level Copy Number Scores")
7. 下载临床数据¶
cldat <- getTCGA_ClinicalData(project = "TCGA-LUAD",save = FALSE,folder = ".",trim = TRUE)
针对的癌症类型:
c("TCGA-READ","TCGA-COAD","TCGA-PAAD","TCGA-ESCA","TCGA-KIRP","TCGA-HNSC",
"TCGA-BLCA","TCGA-STAD","TCGA-CHOL","TCGA-SKCM","TCGA-LUAD","TCGA-LIHC",
"TCGA-KIRC","TCGA-KICH","TCGA-MESO","TCGA-LUSC","TCGA-GBM","TCGA-UVM",
"TCGA-BRCA","TCGA-TGCT","TCGA-THCA")
由于每种癌症类型的临床信息有差异,其他癌症类型,获取临床数据可能会报错,可以通过指定getClinicalData()中的trim = FALSE,返回原始未整理过的数据。
cldat <- getTCGA_ClinicalData(project = "TCGA-LUAD",save = FALSE,folder = ".",trim = FALSE)
8. 过滤表达数据¶
filterGeneTypeExpr()根据某列里面是数据进行过滤,保留filter值的数据。该函数仅适用于getTCGA_RNAseqData获取的count,tpm和fpkm 3个数据框。
expr:表达数据,如:

fil_col:根据那一列进行过滤数据,值为该列的列名。
filter:可选值有"rRNA","misc_RNA","protein_coding","lncRNA","sRNA","snoRNA","miRNA","snRNA","scaRNA","Mt_rRNA"等,具体来说,值应该在所给数据中fil_col列的值中能找到。
rownname:需要作为行名的列,该列的值会去重。
delcol:需要删除的列,为非数值的列的index,默认删除前3列。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD")
expr <- STARdata[["count"]]
table(expr$gene_type)
pc.expr <- filterGeneTypeExpr(expr = expr,
rownname = "gene_name",
fil_col = "gene_type",
filter = FALSE,
delcol = c(1:3))
9.分割数据¶
splitTCGAmatrix(),data的列应该是TCGA病人样本的barcode,参数sample的值为"Tumor"或"Normal",指定sample ="Normal"时,当样本中没有正常样本返回NULL。
turexp <- splitTCGAmatrix(data = expr[,-c(1:3)],sample = "Tumor")
norexp <- splitTCGAmatrix(data = expr[,-c(1:3)],sample = "Normal")
10. 删除重复病人样本¶
delTCGA_dup_sample()函数可以将列为barcode的数据,去除有重复的数据,TCGA数据库的病人有的可能做了几个重复。可以只需要一个。有正常样本和肿瘤样本同时在一个表达矩阵中时,禁用,可以先用splitTCGAmatrix()函数分割正常和肿瘤样本的数据后使用。
expr <- delTCGA_dup_sample(data = pc.expr,col_rename = TRUE)
11. 数据打包下载¶
下载的数据是R对象:
RNAseq:微信公众号生物信息云提供的链接
蛋白组:微信公众号生物信息云提供的链接
TCGA-miRNA_Isoform:微信公众号生物信息云提供的链接
Survival和Phenotype数据(fromUCSC):微信公众号生物信息云提供的链接
临床数据:微信公众号生物信息云提供的链接
12 . 获取某个基因在泛癌中的表达数据¶
geneSymbol是要分析的基因名称的向量;dataType是tpm,fpkm和count中的一种;datafolder来自 getTCGA_RNAseqData()函数下载数据,并存放在某个文件夹中,或者从这里下载(RNAseq:微信公众号生物信息云提供的链接),但这里下载的数据没有fpkm;geneType参照函数filterGeneTypeExpr()中的fil_col,pattern正则表达式匹配datafolder中的数据文件;paired指定是否只获取配对样本的数据;nnorm表示至少包含几个正常样本;得到的数据进行了log2转换。还可以指定projects来选择特定的癌症类型,默认是All,表示获取所有癌症类型的数据,如果想获取其中的几种,其是一个向量,如c("TCGA-LUAD","TCGA-LUSC")
geneSymbol = c("ATG7","ATG12")
datafolder = "G:/DatabaseData/TCGA/new/processedTCGAdata/TCGA-STAR_Exp"
df = getGeneExpData.pancancer(datafolder,
geneSymbol,
geneType = "protein_coding",
dataType = "tpm",
pattern = "STARdata.Rdata$",
projects = "All",
paired = FALSE,
nnorm = 10)
得到的数据样式如下:
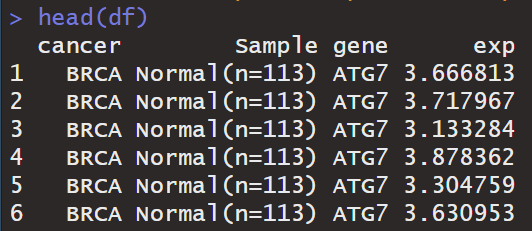
也可以使用:getTCGAgeneExpDat(),该函数不能获取配对样本。
getTCGAgeneExpDat(datafolder,
geneSymbol,
projects = "All",
geneType = "protein_coding"# FALSE
)
13. 单基因在泛癌中表达的箱型图可视化¶
data是由 getGeneExpData.fancancer得到的数据,gene是一个基因,字符串类型;paired表示数据是否是配对样本。
fig <- ggplotGenePancancerExp(data = df,gene= "ATG7",
save = FALSE,folder = ".",paired = FALSE)
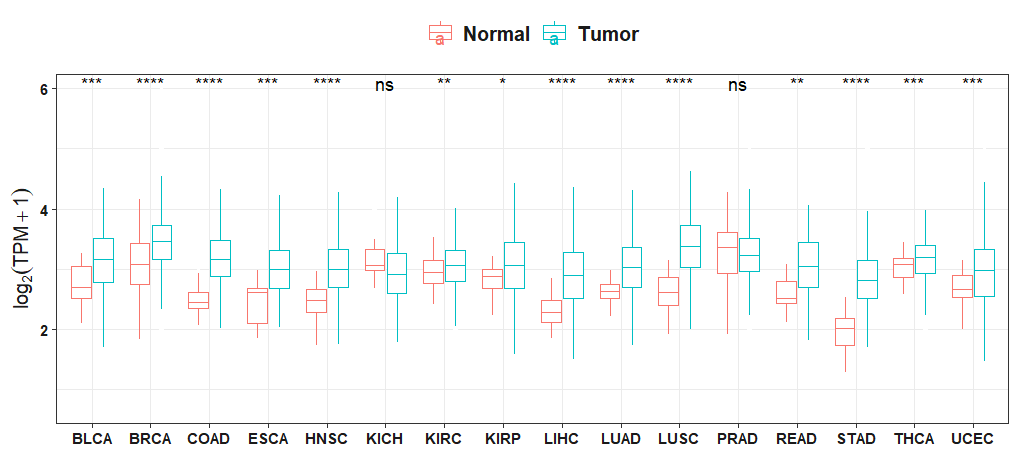
14. 匹配免疫细胞浸润数据与表达数据¶
getInfiltDataOfTCGAsample 函数用于匹配TCGA数据库中的表达数据以及免疫细胞浸润数据,返回一个list(包括匹配后的2个数据框)。免疫数据是从TIMER2数据库下载的数据。
数据下载:【点击下载】。
getInfiltDataOfTCGAsample(expr,idtype = "patient",datatype = "tumor",TIMER2,method)
TIMER2:是下载的免疫细胞浸润数据的文件路径(字符串类型),或者是已经读入该文件的一个数据框。
method:"TIMER","CIBERSORT","CIBERSORT-ABS", "QUANTISEQ","MCPCOUNTER","XCELL","EPIC"中的一种。
expr:是表达数据,行为基因(当然也可以是其他标签),列为样本id。
idtype:"patient" 或 "barcode"中的一个,为"patient" 时会去除重复的样本,并且以病人短id的方式更新数据列名;因为TIMER2下载数据库的id为"TCGA-E7-A6MF-01"这种样式,字符长度为15,如果expr的id长度小于15(通常为"TCGA-E7-A6MF"样式),设置idtype为"barcode",也会被强制使用"patient" 参数;如果expr的id的字符长度大于15(因为TIMER2数据库下载的数据id均为"TCGA-E7-A6MF-01"这种样式),可设置idtype为"barcode",并且数据不会去掉重复病人的数据。
datatype:"tumor"或"normal"中的一个,这取决于expr数据的样本类型,默认为"tumor",有时候我们拿到的expr包含正常样本和肿瘤样本,所以可通过该参数仅匹配肿瘤样本(尽管TIMER2下载的数据包括正常样本,但个人觉得没有太大意义),如果想要正常样本的数据可将该参数设置为"normal"。
15. 融合生存数据与特征数据¶
se <- mergeSurExp(expr
,survival
,survivalFrome = NULL
,Timeunit=1
,TCGA = FALSE
,TCGAfrome = "MedBioInfoCloud"
,feature = NULL
,save = FALSE
,folder = "."
)
该函数主要用于整合TCGA的数据,如果不是TCGA数据库的数据,只需要关注参数expr,survival,save,folder,Timeunit,其他参数不需要考虑,并且,expr行为特征(一般为基因),列为样本;Timeunit的值表示生存时间进行何种转换,Timeunit=1表示不进行任何转换,如果你的生存数据的时间是天,可设置Timeunit=365,转换为年;feature是一个特征子集向量,可以不指定,默认expr的所有行。如果处理TCGA的数据,TCGA应该指定为TRUE,expr应该是getTCGA_RNAseqData()返回结果中的表达数据,如下图:
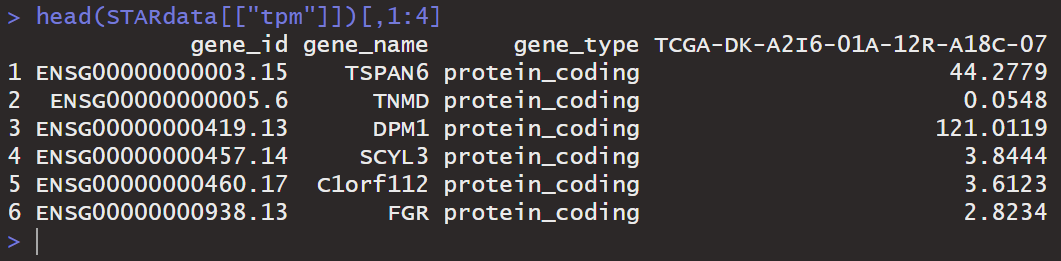
这时,如果生存数据是自己整理的,行名应该和expr的样本一致,或者有交集,如果数据是来自Survival和Phenotype数据(fromUCSC):微信公众号生物信息云提供的链接,可以直接指定survivalFrome = "UCSC2022",数据样式如下:
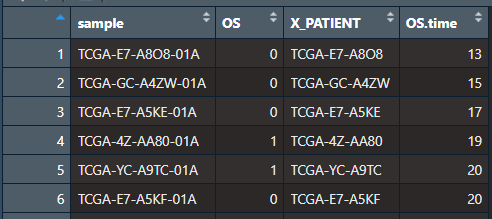
如果从这里下载临床数据:微信公众号生物信息云提供的链接,或是通过本包getClinicalData()函数【trim = TRUE】获取的数据,可以直接指定survivalFrome = "GDCquery_clinic",数据样式如下(至少包含"submitter_id","vitalStat","surTime"):
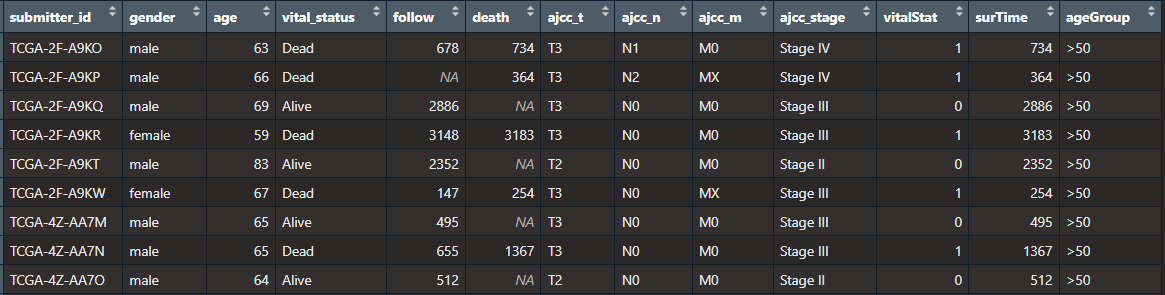
如果指定save = TRUE,会在指定的folder文件夹下保存csv和Rdata的格式文件。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD")
cldat <- getClinicalData(project = "TCGA-LUAD",save = FALSE,folder = ".",trim = TRUE)
se <- mergeSurExp(expr = STARdata[["tpm"]],
survival = cldat,
survivalFrome = "GDCquery_clinic",
TCGA = TRUE)
16.融合表达数据与临床数据¶
mergeClinExp函数与mergeSurExp类似,clinical参数同survival,不过,survivalFrome参数要么为NULL,要么是"UCSC2022"的生存数据(不是表型数据)。其他参数相同。对于TCGA数据来说,mergeSurExp和mergeClinExp都会过滤掉正常样本。
mergeClinExp(expr
,clinical
,survivalFrome = NULL
,TCGA = FALSE
,TCGAfrome = "MedBioInfoCloud"
,feature = NULL
,save = FALSE
,folder = "."
)
17.基因在不同临床分期中的表达分析¶
TCGA_Clin_Analysis(data,clin_feature,feature,title="",width = 5,height = 5, save = TRUE,folder = ".")
data是mergeClinExp函数处理后的的数据,clin_feature为临床特征所在的列的列名。如:ajcc_t","ajcc_n","ajcc_m","ajcc_stage"。
feature:要分析的基因,如:"ATG7"。
title:图片标题。
width,height:图片的宽和高。
3.基于组学数据的评分系统¶
scoringSys()函数用于各种算法的评分,目前支持的方法(参数method)有"ssgsea","gsva","zscore","plage"和"CRDscore",CRDscore方法支持单细胞数据,需要指定study.type = 'scRNAseq'【参考:PMID: 35436363】,study.type有两个值, 'scRNAseq'和'bulk_RNAseq'。"CRDscore"方法不适合小基因集的计算,小基因集容易报错:"Non-enough-overlapping genes to calculate score"。其他方法,也不建议计算小基因集的评分。
expr是一个表达数据,行为基因,列为样本,如果是TCGA数据库的数据,并且是getTCGA_RNAseqData()函数获取的count、tpm,fpkm数据,可以指定TCGA = TRUE,默认为FALSE,需要注意的是,TCGA的数据会自动过滤掉正常样本,只保留肿瘤样本。这里建议使用tpm或者fpkm的数据。
geneset是要分析的基因集,可以是clusterProfiler::read.gmt()函数读入的数据或是GSEABase::getGmt()读入gmt文件的数据。如果自己整理的基因集,建议整理成clusterProfiler::read.gmt()函数读入后的数据形式,也可以是gmt格式的文件路径。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD")
expr <- STARdata[["tpm"]]
score <- scoringSys(expr,
geneset,
TCGA = TRUE,
method ="ssgsea",
study.type = 'bulk_RNAseq',
save = TRUE,folder = ".")
4.WGCNA¶
WGCNA.ModulesPhenotype()函数用于一键式执行WGCNA,输入表型与模块相关性热图,表型特征与模块之间的hub gene。
expr是行为基因,列为样本的TPM表达数据,phenotype是一个表型数据框,行为特征,列为样本,列的顺序应该与expr一致。WGCNA过程中,计算网络相当耗时,如果已经计算过,可以指定recal = FALSE,函数为检测folder文件夹下是否有之前构建网络的数据,如果有会直接加载,不需要重新计算,如果要重新计算,设置为TRUE,colors为热图颜色的向量,至少包含3个梯度色。heatmapH和heatmapW分别指定热图的高和宽,如果最后输入的热图长宽不合适,重新调用此函数,recal = FALSE,重新绘制一次。
outputNet表示是否输出模块内基因之间的网络关系数据,默认TRUE,表示输出,计算时间会很长。
mtc,MM,GS这3个参数用于筛选hub gene的参数,mtc用于指定模块与特征之间的相关性的绝对值,默认0.7,MM,即Module membership,表示 给定基因表达谱与给定Module的eigengene的相关性,是一个基因表达谱与给定模块的Module eigengene之间的相关性度量,它表示了单个基因属于某个特定模块的程度,GS表示基因与特征之间的相关性系数的绝对值。
WGCNA.ModulesPhenotype(expr
,phenotype
,recal = FALSE
,outputNet = TRUE
,colors = blueWhiteRed(50)
,heatmapH=6
,heatmapW=14
,mtc = 0.7
,MM=0.6
,GS=0.6
,folder=".")
4.基于基因集计算评分后执行WGCNA¶
WGCNA.ModulesScoresys()的参数参考WGCNA.ModulesPhenotype()和scoringSys()。
WGCNA.ModulesScoresys(expr
,TCGA = FALSE
,geneset
,method = "ssgsea"
,study.type = 'bulk_RNAseq'
,heatmapH=6
,heatmapW=14
,moduleTraitCor = 0.7
,MM=0.6
,GS=0.6
,folder=".")
expr = STARdata[["tpm"]]
geneset = "G:/myProject/Cytoskeleton/data/geneset/geneset-add-unique.gmt"
folder = "G:/myProject/Cytoskeleton/FBXO22"
WGCNA.ModulesScoresys(expr = expr
,TCGA = TRUE
,geneset = geneset
,folder = folder
)
五.预后模型构建相关函数¶
1.特征选择¶
featureSelect.baseSur()函数可以基于lasso回归,随机森林以及单因素COX回归进行特征选择。
fs <- featureSelect.baseSur(data
,dataFrom = NULL,
feature ="all"
,method = "all"
,cutoff = 0.05
,save = TRUE
,folder = ".")
data是一个数据框,列应该包括生存数据和特征,行为样本。如果数据来源mergeSurExp()函数,前3列的列名应该如下图所示,并且需要指定dataFrom = "mergeSurExp",如果是自己整理的数据,第一列的列名可以随意,但第2列(生存状态)和第3列(生存时间)的列名必须与图中相同。如果没有第一列,将设置dataFrom = NULL。feature,表示要进行分析的特征,默认是所有特征(下图中除了前3列);method的值有lasso、cox、randomForest,单独设置这3个值时,函数返回一个向量(即筛选出的特征),method="all"时,3种方法都执行,最后返回一个list,包括3种方法的结果。cutoff只有当method="cox"或method="all"时被使用。
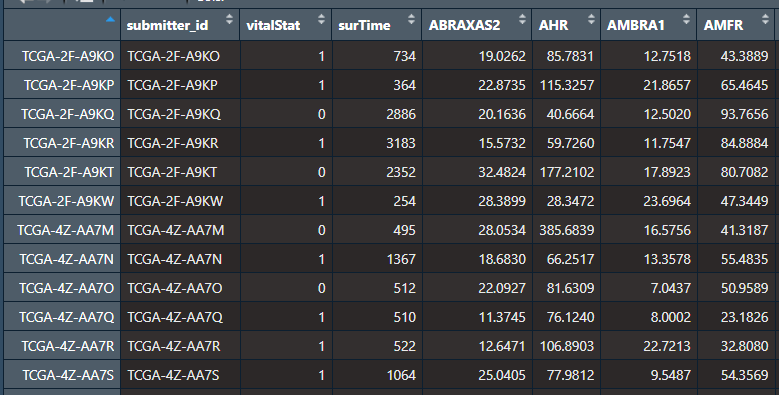
2.多因素COX回归模型¶
MultivariateCOX()函数用于一键式构建多因素COX回归模型。
MultivariateCOX(data
,dataFrom ="mergeSurExp",
feature ="all"
,train_prop = 0.8
,cutoff = 0.05
,save = TRUE
,folder = ".")