MedBioInfoCloud
Functions and data relevant to the tutorials published on the WeChat official account MedBioInfoCloud are encapsulated in this R package. For more learning materials, please refer to BioInfoCloud.
一. Install package¶
This R package has dependencies on several other packages in order to function properly. If you encounter errors during the installation process, it is typically because the required dependencies have not been properly installed. Please install the dependencies individually before proceeding with the installation of the R package.
if (!require("BiocManager", quietly = TRUE))
install.packages("BiocManager")
DependencyPackage <- c("edgeR","DESeq2","TCGAbiolinks")
BiocManager::install(DependencyPackage)
# install.packages("devtools")
devtools::install_github("BioInfoCloud/MedBioInfoCloud")
Some functions with the same parameters are uniformly explained as follows:
save - A boolean value that determines whether the data should be preserved. If save is set to TRUE, the data will be securely stored. Conversely, if it's set to FALSE, the data will remain unsaved.
folder - A string that designates the destination directory for the saved data. This parameter is applicable only when save is set to TRUE. If the specified directory does not exist, it will be automatically created. Kindly note that the folder path should not terminate with a slash ("/"). If the function does not have a save parameter but includes a folder parameter, it indicates that the function will definitely save data, and may not necessarily return valid data. The folder parameter defaults to the current working directory, and it is recommended to set a specific data output directory.
二. TCGA数据库数据挖掘相关函数¶
TCGA数据库首页:GDC Data Portal Homepage (cancer.gov)
1.下载RNAseq数据¶
getTCGA_RNAseqData()返回一个list,包括count,tpm和fpkm 3个数据框。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD",save = TRUE,folder = ".")
2.下载蛋白组数据¶
getTCGA_ProteinExp()返回一个数据框。
Proteome_data <- getTCGA_ProteinExp("TCGA-LUAD",save = TRUE,folder = ".")
3.下载SNV(simple nucleotide variation)数据¶
数据类型为:Masked Somatic Mutation。
snv.dat <- getTCGA_SNV_Masked_data("TCGA-LUAD",save = TRUE,folder = ".")
4.下载miRNA数据¶
参考文章:
https://mp.weixin.qq.com/s/__EjCrJFc08itoF3xqawNg
https://mp.weixin.qq.com/s/-FH0Vi4PaCjhPbEq4-lxbg
https://mp.weixin.qq.com/s/WxgMhwpMAJy_CKTqNdFj0g
(1)Isoform Expression Quantification¶
IsoformEQ <- getTCGA_miRNA_IsoformEQ("TCGA-LUAD",save = TRUE,folder = ".")
(2)miRNA Expression Quantification¶
miRNAEQ <- getTCGA_miRNAEQ("TCGA-LUAD",save = TRUE,folder = ".")
5.下载甲基化数据¶
getTCGA_MethylationData 下载Methylation Beta Value数据。
MetData <- getTCGA_MethylationData("TCGA-LUAD",save = TRUE,folder = ".")
6. 下载CNV(Copy Number Variation)数据¶
getTCGA_CNV.data()函数还在优化中:
cnv.gl <- getTCGA_CNV.data("TCGA-LUAD",save = FALSE,folder = ".",data.type = "Gene Level Copy Number")
cnv.gls <- getTCGA_CNV.data("TCGA-LUAD",save = FALSE,folder = ".",data.type = "Gene Level Copy Number Scores")
7. 下载临床数据¶
cldat <- getTCGA_ClinicalData(project = "TCGA-LUAD",save = FALSE,folder = ".",trim = TRUE)
针对的癌症类型:
c("TCGA-READ","TCGA-COAD","TCGA-PAAD","TCGA-ESCA","TCGA-KIRP","TCGA-HNSC",
"TCGA-BLCA","TCGA-STAD","TCGA-CHOL","TCGA-SKCM","TCGA-LUAD","TCGA-LIHC",
"TCGA-KIRC","TCGA-KICH","TCGA-MESO","TCGA-LUSC","TCGA-GBM","TCGA-UVM",
"TCGA-BRCA","TCGA-TGCT","TCGA-THCA")
由于每种癌症类型的临床信息有差异,其他癌症类型,获取临床数据可能会报错,可以通过指定getClinicalData()中的trim = FALSE,返回原始未整理过的数据。
cldat <- getTCGA_ClinicalData(project = "TCGA-LUAD",save = FALSE,folder = ".",trim = FALSE)
8. 过滤表达数据¶
filterGeneTypeExpr()根据某列里面是数据进行过滤,保留filter值的数据。该函数仅适用于getTCGA_RNAseqData获取的count,tpm和fpkm 3个数据框。
expr:表达数据,如:

fil_col:根据那一列进行过滤数据,值为该列的列名。
filter:可选值有"rRNA","misc_RNA","protein_coding","lncRNA","sRNA","snoRNA","miRNA","snRNA","scaRNA","Mt_rRNA"等,具体来说,值应该在所给数据中fil_col列的值中能找到。
rownname:需要作为行名的列,该列的值会去重。
delcol:需要删除的列,为非数值的列的index,默认删除前3列。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD")
expr <- STARdata[["count"]]
table(expr$gene_type)
pc.expr <- filterGeneTypeExpr(expr = expr,
rownname = "gene_name",
fil_col = "gene_type",
filter = FALSE,
delcol = c(1:3))
9.分割数据¶
splitTCGAmatrix(),data的列应该是TCGA病人样本的barcode,参数sample的值为"Tumor"或"Normal",指定sample ="Normal"时,当样本中没有正常样本返回NULL。
turexp <- splitTCGAmatrix(data = expr[,-c(1:3)],sample = "Tumor")
norexp <- splitTCGAmatrix(data = expr[,-c(1:3)],sample = "Normal")
10. 删除重复病人样本¶
delTCGA_dup_sample()函数可以将列为barcode的数据,去除有重复的数据,TCGA数据库的病人有的可能做了几个重复。可以只需要一个。有正常样本和肿瘤样本同时在一个表达矩阵中时,禁用,可以先用splitTCGAmatrix()函数分割正常和肿瘤样本的数据后使用。
expr <- delTCGA_dup_sample(data = pc.expr,col_rename = TRUE)
11. 数据打包下载¶
下载的数据是R对象:
RNAseq:微信公众号生物信息云提供的链接
蛋白组:微信公众号生物信息云提供的链接
TCGA-miRNA_Isoform:微信公众号生物信息云提供的链接
Survival和Phenotype数据(fromUCSC):微信公众号生物信息云提供的链接
临床数据:微信公众号生物信息云提供的链接
12 . 获取某个基因在泛癌中的表达数据¶
geneSymbol是要分析的基因名称的向量;dataType是tpm,fpkm和count中的一种;datafolder来自 getTCGA_RNAseqData()函数下载数据,并存放在某个文件夹中,或者从这里下载(RNAseq:微信公众号生物信息云提供的链接),但这里下载的数据没有fpkm;geneType参照函数filterGeneTypeExpr()中的fil_col,pattern正则表达式匹配datafolder中的数据文件;paired指定是否只获取配对样本的数据;nnorm表示至少包含几个正常样本;得到的数据进行了log2转换。
geneSymbol = c("ATG7","ATG12")
datafolder = "G:/DatabaseData/TCGA/new/processedTCGAdata/TCGA-STAR_Exp"
df = getGeneExpData.pancancer(datafolder,
geneSymbol,
geneType = "protein_coding",
dataType = "tpm",
pattern = "STARdata.Rdata$",
paired = FALSE,
nnorm = 10)
得到的数据样式如下:
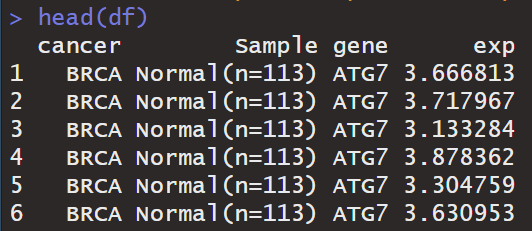
13. 单基因在泛癌中表达的箱型图可视化¶
data是由 getGeneExpData.fancancer得到的数据,gene是一个基因,字符串类型;paired表示数据是否是配对样本。
fig <- ggplotGenePancancerExp(data = df,gene= "ATG7",
save = FALSE,folder = ".",paired = FALSE)
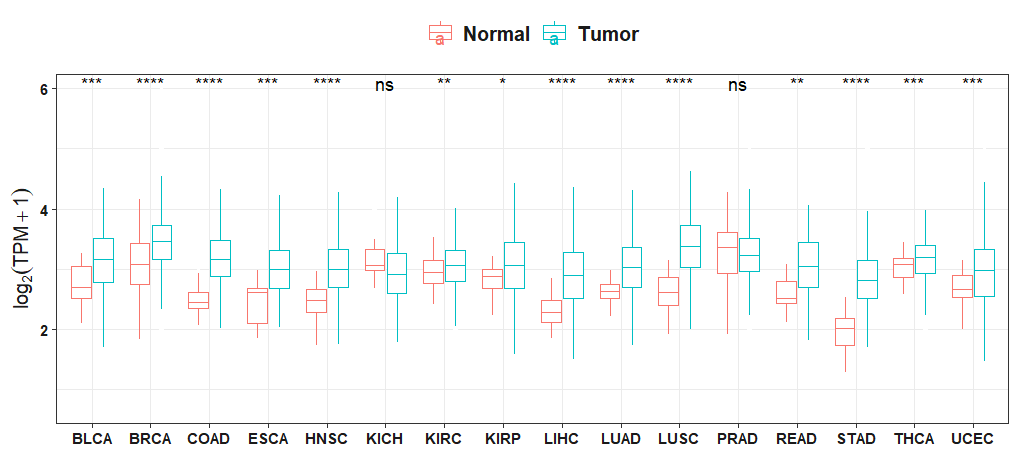
14. 匹配免疫细胞浸润数据与表达数据¶
getInfiltDataOfTCGAsample 函数用于匹配TCGA数据库中的表达数据以及免疫细胞浸润数据,返回一个list(包括匹配后的2个数据框)。免疫数据是从TIMER2数据库下载的数据。
数据下载:【点击下载】。
getInfiltDataOfTCGAsample(expr,idtype = "patient",datatype = "tumor",TIMER2,method)
TIMER2:是下载的免疫细胞浸润数据的文件路径(字符串类型),或者是已经读入该文件的一个数据框。
method:"TIMER","CIBERSORT","CIBERSORT-ABS", "QUANTISEQ","MCPCOUNTER","XCELL","EPIC"中的一种。
expr:是表达数据,行为基因(当然也可以是其他标签),列为样本id。
idtype:"patient" 或 "barcode"中的一个,为"patient" 时会去除重复的样本,并且以病人短id的方式更新数据列名;因为TIMER2下载数据库的id为"TCGA-E7-A6MF-01"这种样式,字符长度为15,如果expr的id长度小于15(通常为"TCGA-E7-A6MF"样式),设置idtype为"barcode",也会被强制使用"patient" 参数;如果expr的id的字符长度大于15(因为TIMER2数据库下载的数据id均为"TCGA-E7-A6MF-01"这种样式),可设置idtype为"barcode",并且数据不会去掉重复病人的数据。
datatype:"tumor"或"normal"中的一个,这取决于expr数据的样本类型,默认为"tumor",有时候我们拿到的expr包含正常样本和肿瘤样本,所以可通过该参数仅匹配肿瘤样本(尽管TIMER2下载的数据包括正常样本,但个人觉得没有太大意义),如果想要正常样本的数据可将该参数设置为"normal"。
15. 融合生存数据与特征数据¶
se <- mergeSurExp(expr
,survival
,survivalFrome = NULL
,Timeunit=1
,TCGA = FALSE
,TCGAfrome = "MedBioInfoCloud"
,feature = NULL
,save = FALSE
,folder = "."
)
该函数主要用于整合TCGA的数据,如果不是TCGA数据库的数据,只需要关注参数expr,survival,save,folder,Timeunit,其他参数不需要考虑,并且,expr行为特征(一般为基因),列为样本;Timeunit的值表示生存时间进行何种转换,Timeunit=1表示不进行任何转换,如果你的生存数据的时间是天,可设置Timeunit=365,转换为年;feature是一个特征子集向量,可以不指定,默认expr的所有行。如果处理TCGA的数据,TCGA应该指定为TRUE,expr应该是getTCGA_RNAseqData()返回结果中的表达数据,如下图:
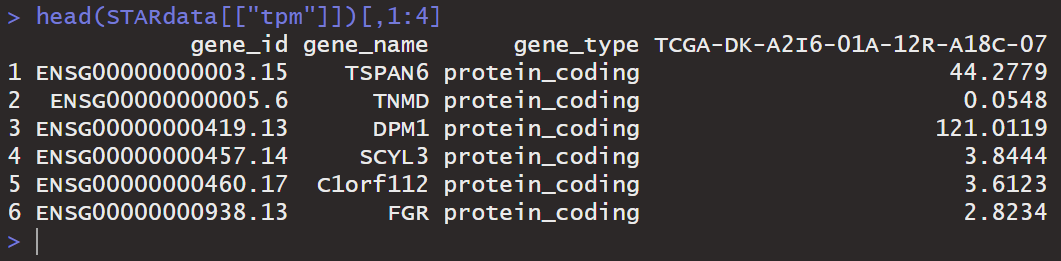
这时,如果生存数据是自己整理的,行名应该和expr的样本一致,或者有交集,如果数据是来自Survival和Phenotype数据(fromUCSC):微信公众号生物信息云提供的链接,可以直接指定survivalFrome = "UCSC2022",数据样式如下:
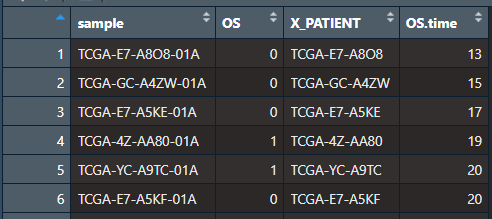
如果从这里下载临床数据:微信公众号生物信息云提供的链接,或是通过本包getClinicalData()函数【trim = TRUE】获取的数据,可以直接指定survivalFrome = "GDCquery_clinic",数据样式如下(至少包含"submitter_id","vitalStat","surTime"):
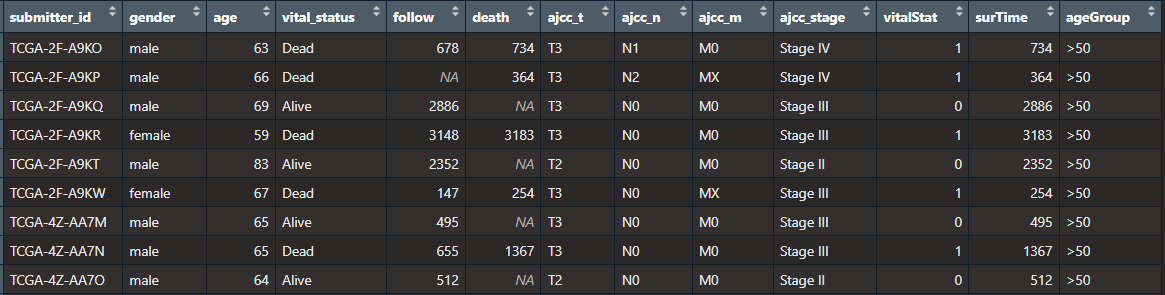
如果指定save = TRUE,会在指定的folder文件夹下保存csv和Rdata的格式文件。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD")
cldat <- getClinicalData(project = "TCGA-LUAD",save = FALSE,folder = ".",trim = TRUE)
se <- mergeSurExp(expr = STARdata[["tpm"]],
survival = cldat,
survivalFrome = "GDCquery_clinic",
TCGA = TRUE)
16.融合表达数据与临床数据¶
mergeClinExp函数与mergeSurExp类似,clinical参数同survival,不过,survivalFrome参数要么为NULL,要么是"UCSC2022"的生存数据(不是表型数据)。其他参数相同。对于TCGA数据来说,mergeSurExp和mergeClinExp都会过滤掉正常样本。
mergeClinExp(expr
,clinical
,survivalFrome = NULL
,TCGA = FALSE
,TCGAfrome = "MedBioInfoCloud"
,feature = NULL
,save = FALSE
,folder = "."
)
17.基因在不同临床分期中的表达分析¶
TCGA_Clin_Analysis(data,clin_feature,feature,title="",width = 5,height = 5, save = TRUE,folder = ".")
data是mergeClinExp函数处理后的的数据,clin_feature为临床特征所在的列的列名。如:ajcc_t","ajcc_n","ajcc_m","ajcc_stage"。
feature:要分析的基因,如:"ATG7"。
title:图片标题。
width,height:图片的宽和高。
三.一些数据处理和分析过程中的基础函数¶
1.输出gmt文件¶
outputGmtFile()函数中description默认为NA,如果指定,应该是一个长度与input相同,用于描述每个基因集的字符串向量。filename应该是一个.gmt结尾的文件名称,可包括路径。input是一个list或是一个data.frame,如果是list,list中每一个对象是一个向量(基因),每一个对象应该有一个合适的名称,相当于基因集的名称,下面是一个input接收list数据对象案例:
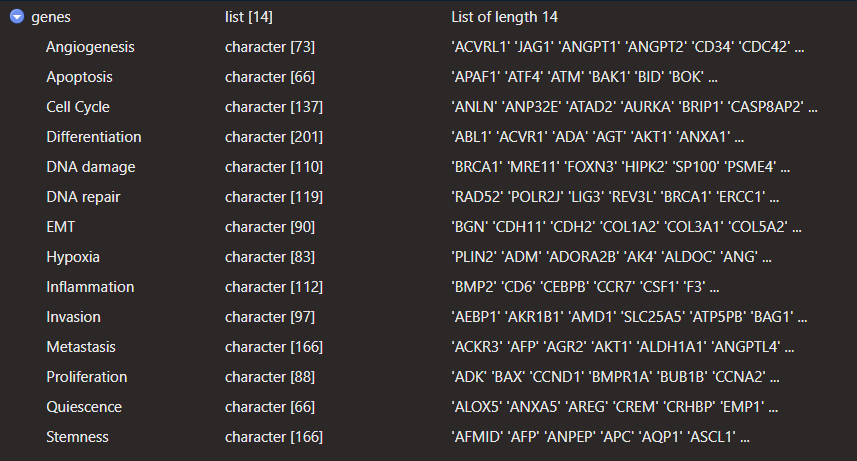
outputGmtFile(input = genes,description = NA,filename = "./gs.gmt")
如果input是一个数据框,应该包括两列,第一列是关于基因集描述的术语,第二列是基因,这时,如果需要指定description的值,长度应该等于input第一列值作为集合的长度。下面是一个input输入作为数据框的案例:
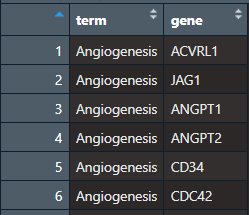
outputGmtFile(input = cg,description = NA,filename = "./gs.gmt")
2.将一个基因集生成一个gmt文件¶
geneset2gmt(geneset,genesetname,description = NA,return = "data.frame",folder= ".",filename)
geneset是一个向量,向量的每一个值应该是一个基因名称,也可以是以.txt结尾的文件路径(文件内容应该是一行一个基因名称),genesetname为这个基因集取的名字,需要指定;return的值为"data.frame"或者是"GeneSetCollection",表示什么样的值,相当于分别用clusterProfiler::read.gmt()函数和GSEABase::getGmt()函数读入gmt文件的结果,可以根据后续需要选择返回结果;该函数内部调用outputGmtFile,其他参数同outputGmtFile()。
3.整理gmt文件¶
tidy.gmt()用于统计或者合并多个gtf文件。filepath是文件路径,不用指定到具体文件,函数会自动匹配文件夹下所有以".gmt"格式结尾的文件,当然,filepath也可以是一个包括多个gmt格式文件完整路径的向量,比如:c("./gs1.gmt","/home/user/data/gs2.gmt","/home/user/geneset/gs3.gmt");fun的值是"stat" 或 "merge"中的一个,"stat"表示统计每个gmt文件中的基因个数,并输入一个csv文件,"merge"就是合并多个基因集,会返回一个数据框(不做统计,相当于函数clusterProfiler::read.gmt()的读入结果),同时输出一个gmt格式文件,无论那个,都会输出一个.unique.txt的文件以记录所有基因集中的文件;termName用于指定当fun = "stat" 时,所有基因集的统一名称,相当于列名,默认NULL(输入的列名是trem);Source是所有基因集的来源,默认"",长度应该为1;filename可以不指定文件后缀名,指定后缀名,输出也会根据fun的不同而添加相应的后缀名。addTotal的值为TRUE或FALSE,只有fun = "merge"时被使用,表示是否将所有基因集合并作为一个新的基因,相当于在输出的gmt文件中多了一行,此时作为基因集的名称由termName指定,如果termName为NULL,那么新组合的基因集名称为Total gene。
filepath <- "G:/publicData/base_files/GeneSet/Cytoskeleton/"
folder <- "G:/myProject/data/geneset"
gsdf <- tidy.gmt(filepath
,fun = "stat"
,Source = ""
,addTotal = TRUE
,termName=NULL
,save = TRUE
,folder = folder
,filename = "geneset"
)
4.整理Reactome数据框下载的通路信息文件¶
tidyGene.fromeReactome()与tidy.gmt()类似。Source默认为"Reactome";需要注意的是,下载的数据文件名以通路名称[通路ID]的形式命名,文件格式为tsv。fun的值是"stat" 时返回数据比tidy.gmt()多了PathwayID列;fun = "merge"时,2者输出一样。其他参数同tidy.gmt()。
filepath <- "G:/publicData/base_files/GeneSet/DNA_Repair"
folder = "G:/myProject/DNA_Repair"
df <- tidyGene.fromeReactome(filepath = filepath
,fun = "stat"
,Source = "Reactome"
,addTotal = TRUE
,termName=NULL
,save = TRUE
,folder =folder
,filename = "geneset")
5.从基因组文件gtf中提取基因相关信息¶
gtf参数是一个gtf文件路径。
(1)提取基因长度信息¶
gtf_file <- "path/to/your/file.gtf" # 替换为实际的GTF文件路径
gene_lengths <- getGeneLenFromeGTF(gtf_file)
(2)提取基因类型信息¶
genetype <- getGeneTypeInfoFromeGTF(gtf_file)
(3)提取基因的基础信息(长度 + 类型)¶
geneinfo <- getGeneBaseInfo(gtf)
6.RNAseq数据转换¶
参考:RNAseq数据分析中count、FPKM和TPM之间的转换 (qq.com)
函数RNAseqDataTrans():
RNAseqDataTrans(data,tfun,species,gtype)
data是RNAseq数据,行可以是 'Ensemble' or 'Symbol'类型,列为样本;
tfun must be one of "Counts2TPM", "Counts2FPKM", or "FPKM2TPM";
species的值可以是 "hsa"、"mus";
gtype must be one of 'Ensemble' or 'Symbol'.
类似的函数RNAseqDataConversion
data <- RNAseqDataConversion(data,type,species = "homo",gtf = NULL)
data是RNAseq数据,行为基因(symbol),列为样本;type must be one of "Counts2TPM", "Counts2FPKM", or "FPKM2TPM".
species的值可以是 "homo"、"mus",或者是NULL。如果你的数据是人或者小鼠,只需要指定为 "homo"或"mus",如果不是,species设置为NULL,同时提供gtf格式的参考基因组文件。
7.基因集对象转换¶
read.gmt.to.getGmt()实现clusterProfiler::read.gmt()函数读入的数据转换为GeneSetCollection对象【相当于GSEABase::getGmt()读入gmt文件的结果】。genesetdf是一个数据框,有两列数据(term和gene)。
gs <- read.gmt.to.getGmt(genesetdf)
GeneSetCollection.to.df()函数相反,将GeneSetCollection对象转换为一个数据框:
gsdf <- GeneSetCollection.to.df(GeneSetCollection)
四.基础分析相关函数的使用¶
1.差异表达分析¶
data为表达数据,行为基因名称,列为样本名称,group是一个数据框,只有一列为group的值,其值是二分类的字符串标签(如:Tumor,Normal),行名为样本名称,其顺序与data的列名一致。comparison是一个由group中的二分类标签值用-链接,如"Tumor-Normal",表示Tumor组与Normal进行差异表达分析。method是DESeq2, edgeR和 limma中的一种,RNAseq数据建议使用DESeq2或edgeR,芯片数据使用limma。filter是否过滤数据,默认为TRUE。注意:该函数值适合count数据。cutFC和 cutFDR是筛选差异表达基因的条件。
DEG <- geneDEAnalysis(data, group, comparison,method = "DESeq2", cutFC = 2,cutFDR = 0.05,filter = TRUE)
标准化后的芯片数据可用arrayDataDEA_limma函数:
group是一个长度和data列数一样的向量,其中每一个元素值应该对应样本的分组,且唯一值只有2种,comparison同上。
comparison = "Liver_Metastasis-Primary" # 必须在函数外定义
DEG = arrayDataDEA_limma(data = Liver_M_vs_Primary,
group = rep(c("Liver_Metastasis","Primary"),
times = c(length(LiverID),length(PrimaryID))),
comparison = comparison,
cutFC = 2,cutFDR = 0.05
)
2. 火山图可视化¶
plotDEGvolcanoFig函数用于差异表达分析的火山图可视化,该函数在进行绘制前,差异分析需要进行一些处理。
plotDEGvolcanoFig <- function(data,x,y,cut_pvalue,cutFC,title,group,colour,label)
data是差异分析的结果,x是x轴的列(log2FC),y轴是p-value/FDR,cut_pvalue是显著性的截断值,cutFC是log2FC绝对值的绝对值。title是标题,group是分组所在的列,colour是颜色,长度应该以group的唯一值相同,label是需要显示的基因名所在的列。
2.基于组学数据的评分系统¶
scoringSys()函数用于各种算法的评分,目前支持的方法(参数method)有"ssgsea","gsva","zscore","plage"和"CRDscore",CRDscore方法支持单细胞数据,需要指定study.type = 'scRNAseq'【参考:PMID: 35436363】,study.type有两个值, 'scRNAseq'和'bulk_RNAseq'。"CRDscore"方法不适合小基因集的计算,小基因集容易报错:"Non-enough-overlapping genes to calculate score"。其他方法,也不建议计算小基因集的评分。
expr是一个表达数据,行为基因,列为样本,如果是TCGA数据库的数据,并且是getTCGA_RNAseqData()函数获取的count、tpm,fpkm数据,可以指定TCGA = TRUE,默认为FALSE,需要注意的是,TCGA的数据会自动过滤掉正常样本,只保留肿瘤样本。这里建议使用tpm或者fpkm的数据。
geneset是要分析的基因集,可以是clusterProfiler::read.gmt()函数读入的数据或是GSEABase::getGmt()读入gmt文件的数据。如果自己整理的基因集,建议整理成clusterProfiler::read.gmt()函数读入后的数据形式,也可以是gmt格式的文件路径。
STARdata <- getTCGA_RNAseqData("TCGA-LUAD")
expr <- STARdata[["tpm"]]
score <- scoringSys(expr,
geneset,
TCGA = TRUE,
method ="ssgsea",
study.type = 'bulk_RNAseq',
save = TRUE,folder = ".")
3.WGCNA¶
WGCNA.ModulesPhenotype()函数用于一键式执行WGCNA,输入表型与模块相关性热图,表型特征与模块之间的hub gene。
expr是行为基因,列为样本的TPM表达数据,phenotype是一个表型数据框,行为特征,列为样本,列的顺序应该与expr一致。WGCNA过程中,计算网络相当耗时,如果已经计算过,可以指定recal = FALSE,函数为检测folder文件夹下是否有之前构建网络的数据,如果有会直接加载,不需要重新计算,如果要重新计算,设置为TRUE,colors为热图颜色的向量,至少包含3个梯度色。heatmapH和heatmapW分别指定热图的高和宽,如果最后输入的热图长宽不合适,重新调用此函数,recal = FALSE,重新绘制一次。
outputNet表示是否输出模块内基因之间的网络关系数据,默认TRUE,表示输出,计算时间会很长。
mtc,MM,GS这3个参数用于筛选hub gene的参数,mtc用于指定模块与特征之间的相关性的绝对值,默认0.7,MM,即Module membership,表示 给定基因表达谱与给定Module的eigengene的相关性,是一个基因表达谱与给定模块的Module eigengene之间的相关性度量,它表示了单个基因属于某个特定模块的程度,GS表示基因与特征之间的相关性系数的绝对值。
WGCNA.ModulesPhenotype(expr
,phenotype
,recal = FALSE
,outputNet = TRUE
,colors = blueWhiteRed(50)
,heatmapH=6
,heatmapW=14
,mtc = 0.7
,MM=0.6
,GS=0.6
,folder=".")
4.基于基因集计算评分后执行WGCNA¶
WGCNA.ModulesScoresys()的参数参考WGCNA.ModulesPhenotype()和scoringSys()。
WGCNA.ModulesScoresys(expr
,TCGA = FALSE
,geneset
,method = "ssgsea"
,study.type = 'bulk_RNAseq'
,heatmapH=6
,heatmapW=14
,moduleTraitCor = 0.7
,MM=0.6
,GS=0.6
,folder=".")
expr = STARdata[["tpm"]]
geneset = "G:/myProject/Cytoskeleton/data/geneset/geneset-add-unique.gmt"
folder = "G:/myProject/Cytoskeleton/FBXO22"
WGCNA.ModulesScoresys(expr = expr
,TCGA = TRUE
,geneset = geneset
,folder = folder
)
五.预后模型构建相关函数¶
1.特征选择¶
featureSelect.baseSur()函数可以基于lasso回归,随机森林以及单因素COX回归进行特征选择。
fs <- featureSelect.baseSur(data
,dataFrom = NULL,
feature ="all"
,method = "all"
,cutoff = 0.05
,save = TRUE
,folder = ".")
data是一个数据框,列应该包括生存数据和特征,行为样本。如果数据来源mergeSurExp()函数,前3列的列名应该如下图所示,并且需要指定dataFrom = "mergeSurExp",如果是自己整理的数据,第一列的列名可以随意,但第2列(生存状态)和第3列(生存时间)的列名必须与图中相同。如果没有第一列,将设置dataFrom = NULL。feature,表示要进行分析的特征,默认是所有特征(下图中除了前3列);method的值有lasso、cox、randomForest,单独设置这3个值时,函数返回一个向量(即筛选出的特征),method="all"时,3种方法都执行,最后返回一个list,包括3种方法的结果。cutoff只有当method="cox"或method="all"时被使用。
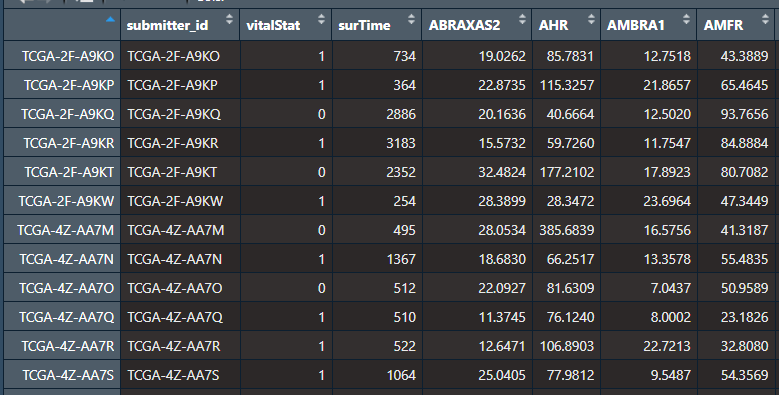
2.多因素COX回归模型¶
MultivariateCOX()函数用于一键式构建多因素COX回归模型。
MultivariateCOX(data
,dataFrom ="mergeSurExp",
feature ="all"
,train_prop = 0.8
,cutoff = 0.05
,save = TRUE
,folder = ".")