细胞注释-SingleR
单细胞转录组的结果其实就是基因和细胞的矩阵,基于此数据可以做PCA、tSNE、差异分析等,那么已有的Seurat工具便可做此类分析,进行数据的可视化。但是聚类出来的细胞类型我们是不清楚的,只知道分类而已,这是没有意义的。
在定义细胞类型之前,需要确定就哪种聚类结果来做,是图聚类的结果还是k-means某一类的结果。如何来确定?看看分群的tsne图是一个不错的参考。
一般识别细胞类型有三种方法:
-
从文献中获取已经验证的Marker
-
根据Marker基因,采用CellMarker或者panglaoDB数据库,进行细胞注释,可以采用超几何分布算法来进行精确性验证
-
采用一些自动化注释的软件,例如SingleR, Garnett, celaref等
目前主流的方法是利用CellMarker等数据库进行注释。这些数据库其实就是一个细胞类型和marker基因表。根据marker基因列表与分析出来的差异基因列表以及基因丰度的关系(往往是做一个热图)来推断某个cluster属于哪一种细胞类型。最准确可靠的还是追踪文献,但这个需要大量的经验和追踪热点的精力。
SingleR是一个R包,是单细胞数据分析中细胞注释工具,它可以根据已有的参考数据集对单细胞数据进行自动注释,并且能够与Seurat工具结合,直接使用Seurat的结果作为输入数据,简单快捷。
一.Seurat包原理¶
Step1:建立Marker基因集合¶
根据参考数据集,我们得到Marker基因。主要方法是默认基于差异倍数(其他比如秩和检验或者T检验)。主要分2 步:
1.1 计算差异倍数¶
以计算参考集A类细胞的Marker基因为例,先计算得到参考集中各个基因在A类细胞和其他细胞中表达量的中位数(因为参考集,每种细胞都有大量重复,所以可以计算中位数),然后根据中位数将参考集中细胞类型进行两两比较,计算表达差异倍数。
SingleR自带5个人的参考数据库和2个小鼠的参考数据库,,每个数据库导入的方法如下:
library(SingleR)
library(Seurat)
# human
hpca.se <- HumanPrimaryCellAtlasData()
bpe.se <- BlueprintEncodeData()
DICE <- DatabaseImmuneCellExpressionData()
NHD <- NovershternHematopoieticData()
MID <- MonacoImmuneData()
# mouse
MRD <- MouseRNAseqData()
IGD <- ImmGenData()
第一次调用需要下载到本地,后续调用直接加载本地文件,但第一次下载过程比较耗费时间,而且国内网络不稳定可能会失败。
Mouse:
-
Immunological Genome Project (ImmGen): 收集了830个微阵列样本,我们将其分类为20个主要细胞类型,进一步注释为253个子类型 (Heng et al. 2008)。The ImmGen reference consists of microarray profiles of puremouse immune cells from the project of the same name (Heng et al. 2008). This is currently the most highly resolved immune reference- possibly overwhelmingly so, given the granularity of the fine labels。
-
MRD参考包含从基因表达综合库下载的小鼠大体积rna序列数据集(Benayoun等人,2019年)。有各种各样的细胞类型,同样主要来自血液,但也包括其他一些组织。
Human:
- Human Primary Cell Atlas (HPCA): 基因表达集合(GEO数据集),包含713个微阵列样本,分类为38种主要细胞类型,进一步注释为169个子类型 (Mabbott et al. 2013)。大多数标签指的是血液亚群,但细胞类型从其他组织也可用。
- Blueprint/ENCODE 参考由 Blueprint (Martens and Stunnenberg 2013)和 ENCODE 项目(ENCODE Project Consortium 2012)产生的纯基质和免疫细胞的大量 RNA-seq 数据组成。
- DICE参考由来自同名项目(Schmiedel等人,2018年)的排序细胞群的大量RNA-seq样本组成。
- Novershtern参考资料(以前称为分化图)由GSE24759中排序的造血细胞群的微阵列数据集组成(Novershtern et al. 2011)。
- Monaco参考由来自GSE107011的免疫细胞群的大量RNA-seq样本组成(Monaco et al. 2019)。
1.2 确定Marker的数目¶
选取差异倍数最高的N个基因(upregulated)作为A类细胞的marke基因,那么A类细胞最终的Marker基因是两两比较差异基因的并集。这个指标N可以根据项目的情况优化调整。
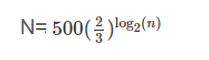
Tips:
-
Marker基因的计算方法可以自己定义,比如定义为pairwise binomial tests
-
可以自己提供Marker基因list给到SingleR
Step2:计算待注释的细胞与参考集中已知细胞表达量的斯皮尔曼相关性¶
用于计算相关性的基因为对应类型的Marker基因,这样可以减少非marker基因的随机噪音,提高结果的特异性。
SingleR计算相关性有两种方法:
- sd—— 参考数据集中所有样本的标准差超过阈值的基因。我们选择阈值,从3000-4000个基因开始。
- de——与其他细胞类型相比,在一种细胞类型中有较高中位数表达的前N个基因。我们使用可变的N,这取决于分析中使用的细胞类型的数量(K)。
计算待测细胞X与参考集A类细胞的相关系数,细胞X与参考集A类细胞的相关系数为80%分位数(由于参考集A类细胞有很多重复,会得到多个相关系数)。也就是细胞X与参考集中的每一类细胞只有1个相关系数,这就避免了参考库中数据异质性导致的误差。
Step3:细胞注释¶
3.1 首先获得第一轮的细胞注释结果,分以下2种结果:
-
待测细胞X类型确定,相关系数在2种类型细胞中最高的;
-
待测细胞X 与已知A类、C类、D类等相关系数都很较高且都差不多(0.39~0.4),无法确定细胞类型。
3.2 进行新一轮细胞注释(微调)
对于上述2)的情况,singleR按照如下方法进行进一步注释:
(1)先singleR使用“淘汰原则”来逐步缩小范围,而非要求一步到位。在完成第一步相关系数计算后,先淘汰相关系数最小或者与相关系数最高值小0.05以上的细胞类型。
(2)基于剩余的细胞类型重新构建参考集,回到以上的Step1进行新一轮的Marker基因筛选以及相关系数判断。直到最后一轮比拼只在两种细胞中开展,SingleR软件才会选择参考集中相关系数更高的那个细胞类型作为细胞X的注释结果。
这种方法的优势在于在每次循环判定过程中,每类细胞的Marker基因都是重新计算的,这能够逐步提高对参考集中高度相似的细胞类型的区分能力,最终得出一个稳定可靠的注释结果。
Step4:注释结果诊断¶
4.1 基于细胞得分进行诊断¶
最明显的诊断方法是根据每个待注释细胞的得分,该得分就是我们之前计算的相关性值。对于注释结果比较清晰的细胞来说,这些细胞在一个标签的得分很显著的高于其他标签得分的,这也是我们所期望的结果。
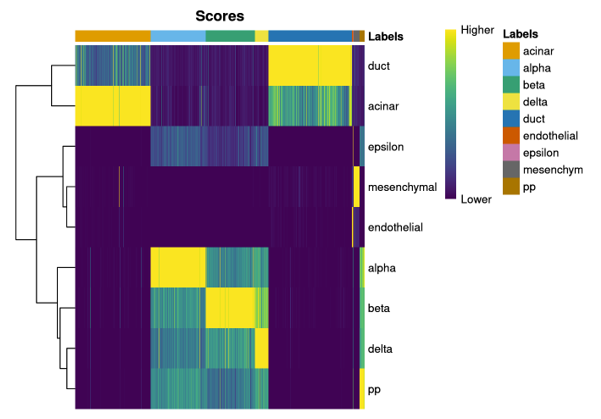
基于得分的细胞注释结果热图
注:每一列是一个细胞,每一行为参考集里的细胞类型标签,每一格表示细胞在该标签获得的得分。颜色代表得分高低。
4.2 基于细胞delta值进行诊断¶
我们使用待注释细胞的delta值识别低质量注释结果或者模棱两可的注释结果。Delta值的定义是每个细胞的注释标签的得分与所有标签得分的中位数的差值。Delta值低,说明注释结果不明确。
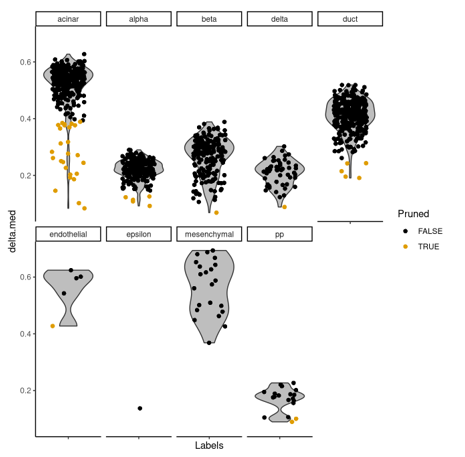
基于delta值细胞分布
注:每一格子图表示一个细胞类型,子图里每个点表示一个细胞。横坐标为分配到该类型的细胞,纵坐标为该细胞的 delta中位数。
4.3基于细胞marker基因进行诊断¶
检查测试数据集中每个标记的marker基因(上调)的表达情况,如果测试数据集中的某个细胞确信地分配给了特定标签,我们希望标记为该标签的marker基因具有很高的表达。且具有生物学意义。相比得分和delta诊断方法,该方法更可解释的诊断可视化。但对于大量标签可能不太适用,还有通过热图无法判断相近标签注释的准确性。
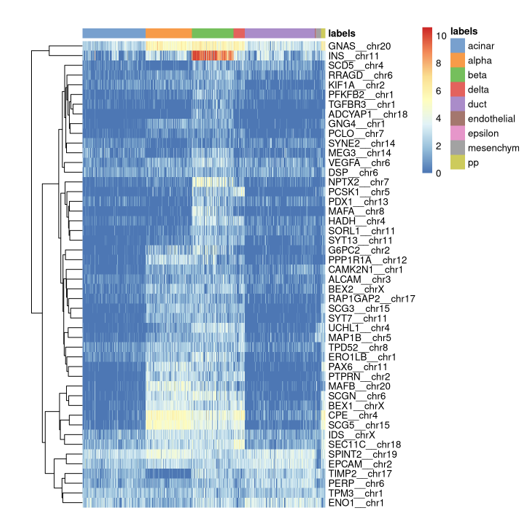
marker基因表达热图
注:每一行为一个基因,每一列为细胞,颜色表示基因表达量。labels为细胞的注释结果。
4.4 与无监督的聚类结果进行比较¶
最后一个诊断方法是将注释结果与无监督聚类结果进行比较。我们期望的是注释结果与无监督聚类结果是一致的。
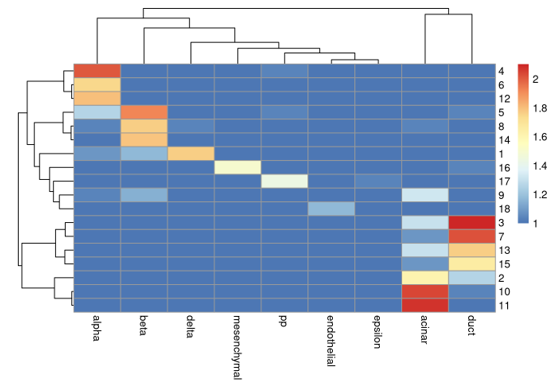
每个注释结果及cluster的细胞数的热图
注:每行为聚类编号,每列为注释结果标签,每一格子是每类细胞被分配到每个标签的细胞数取log
上面内容参考:https://mp.weixin.qq.com/s/_a0JoPIdBWK6DwYke0KNEA
二.使用案例¶
1.加载Seurat对象数据¶
使用上次分析保存的数据:https://mp.weixin.qq.com/s/YUcPWEZ8UvwgaqL4qrLHPA
library(SingleR)
library(Seurat)
load('data/sce.output.merge.GSE130001.Rdata')
2.加载参考数据¶
我们这里的数据是人的。
hpca.se <- HumanPrimaryCellAtlasData()
hpca.se
3.开始注释¶
注释结果是labels列。这里注意一下,如果不写参数clusters = clusters,那么程序是对每个细胞单独注释而不是对类群,对每个细胞注释耗时特别长,如果想快速出结果,就对类群注释,想看仔细一点就对单个细胞注释。
同时指定HumanPrimaryCellAtlasData作为注释集
sce1 <- sce ###赋值给其他变量,避免修改原变量。
sce_for_SingleR <- GetAssayData(sce1, slot="data")
sce_for_SingleR
clusters <- sce1@meta.data$seurat_clusters
pred.hesc <- SingleR(test = sce_for_SingleR,
ref = hpca.se,
labels = hpca.se$label.main,
method = "cluster",
clusters = clusters,
assay.type.test = "logcounts",
assay.type.ref = "logcounts")
table(pred.hesc$labels)
MedBioInfoCloud: table(pred.hesc$labels)
Chondrocytes Endothelial_cells Epithelial_cells NK_cell Tissue_stem_cells
1 1 9 1 1
可视化注释结果:
celltype = data.frame(ClusterID=rownames(pred.hesc),
celltype=pred.hesc$labels,
stringsAsFactors = F)
sce1@meta.data$singleR = celltype[match(clusters,celltype$ClusterID),'celltype']
P9 <- DimPlot(sce1, reduction = "tsne", group.by = "singleR")
P9

直接修改原来聚类的名称,这里类似于手动注释细胞类型:
sce2 <- sce
head(celltype)
new.cluster.ids <- celltype$celltype
names(new.cluster.ids) <- levels(sce2)
sce2 <- RenameIdents(sce2, new.cluster.ids)
P10 <- DimPlot(sce2, reduction = "tsne", label = TRUE, pt.size = 0.5) + NoLegend()
P9 + P10
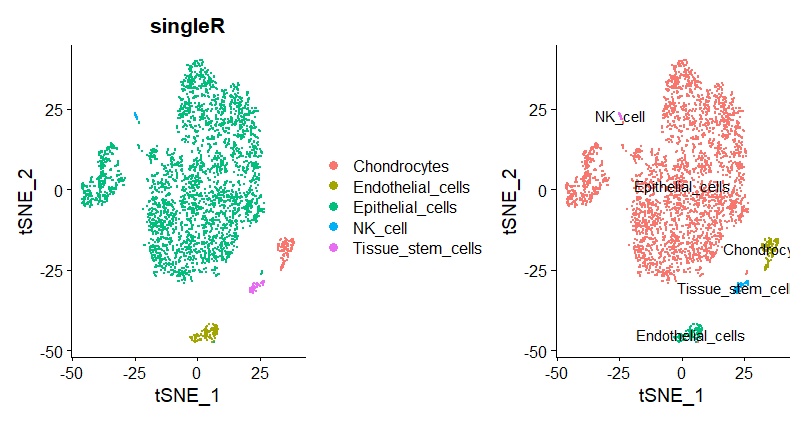
不过,很神奇,我们这里的样本是CD45阴性的细胞,按道理是不会有NK细胞的,但是这里有些细胞注释为NK细胞,是有问题的。
# 标志物
# immune (CD45+,PTPRC),
# epithelial/cancer (EpCAM+,EPCAM),
# stromal (CD10+,MME,fibo or CD31+,PECAM1,endo)
当然,原文献只选择那些通过原始质量控制和显示零CD45读取计数的细胞。我这里演示并没有去除CD45不表达的细胞【尽管流式筛了一边,偶尔还会有些漏网之鱼】,有可能是这个原因。但是作者的分析中仍然存在一些CD45读取计数为零的免疫细胞,他的解释是这可能反映了缺失事件或给定有限的测序深度的采样伪影。这里只做演示,具体需要根据知识背景判断。
MedBioInfoCloud: phe=sce@meta.data
MedBioInfoCloud: table(phe$singleR)
Chondrocytes Endothelial_cells Epithelial_cells NK_cell Tissue_stem_cells
143 141 3642 26 70
4.细胞注释的可靠性¶
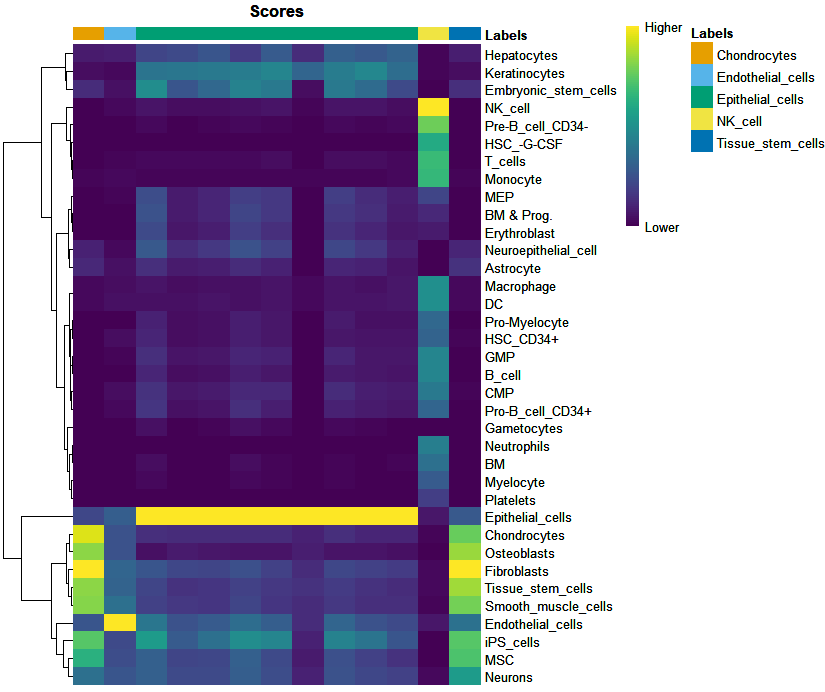
plotScoreHeatmap显示所有引用细胞类型上所有细胞的得分,这允许用户检查数据集中预测细胞类型的可信度。每个类群/细胞的实际分配标签显示在顶部的颜色栏中。关键点是检查分数(scores)在每个类群/细胞中的分布情况。理想情况下,每个类群/细胞(即热图的列)应该有一个明显大于其他细胞的分数,这表明它明确地分配给了单个标签。
plotScoreHeatmap(pred.hesc)
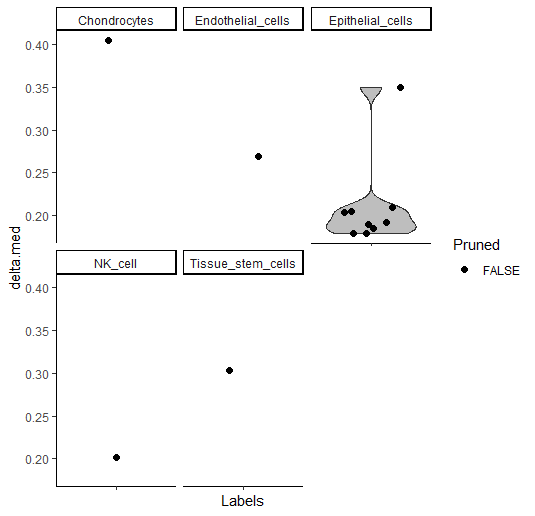
另外还可以使用plotDeltaDistribution查看注释的可靠性,deltas值越小说明可靠性越低。
# 基于 per-cell “deltas”诊断,Delta值低,说明注释结果不是很明确
plotDeltaDistribution(pred.hesc, ncol = 3)
5.自定义注释数据集¶
我们可以自己下载别人注释好的表达矩阵数据来注释我们的数据,这里我们用前面注释的数据自己为自己做参考来介绍怎么自定义注释数据集:
myref <- sce2##这里为了检测,我们将参考数据集与目标数据集用同一个数据进行测试
myref$celltype <- Idents(myref)
table(Idents(myref))
# 读入参考数据集 -------
Refassay <- log1p(AverageExpression(myref, verbose = FALSE)$RNA)#求细胞的平均表达
#Ref <- textshape::column_to_rownames(Ref, loc = 1)#另一种得到参考矩阵的办法
head(Refassay)#看看表达矩阵长啥样
其实,你需要准备的就是一个细胞的表达矩阵,类似于下面这样的:
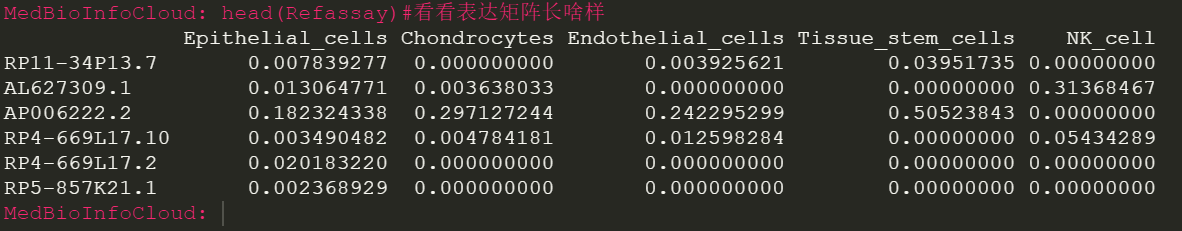
然后,使用参考注释数据集构建一个SingleCellExperiment对象。
ref_sce <- SingleCellExperiment::SingleCellExperiment(assays=list(counts=Refassay))
#参考数据集需要构建成一个SingleCellExperiment对象
# if(!require(scater))BiocManager::install('scater')
library(scater)
ref_sce <- scater::logNormCounts(ref_sce)
colData(ref_sce)$Type <- colnames(Refassay)
###提取自己的单细胞矩阵##########
testdata <- GetAssayData(sce, slot="data")
pred <- SingleR(test=testdata,
ref=ref_sce,
labels=ref_sce$Type,
#clusters = scRNA@active.ident
)
table(pred$labels)
cellType=data.frame(seurat= sce@meta.data$seurat_clusters,
predict=pred$labels)#得到seurat中编号与预测标签之间的关系
lalala <- as.data.frame(table(cellType[,1:2]))
finalmap <- lalala %>% group_by(seurat) %>% top_n(n = 1, wt = Freq)#找出每种seurat_cluster注释比例最高的对应类型
finalmap <-finalmap[order(finalmap$seurat),]$predict#找到seurat中0:12的对应预测细胞类型
print(finalmap)
testname <- sce
new.cluster.ids <- as.character(finalmap)
names(new.cluster.ids) <- levels(testname)
testname <- RenameIdents(testname, new.cluster.ids)
P11 <- DimPlot(sce,label = T)
P12 <- DimPlot(testname,label = T)#比较一下测试数据与参考数据集之间有没有偏差
P11 + P12 + P9
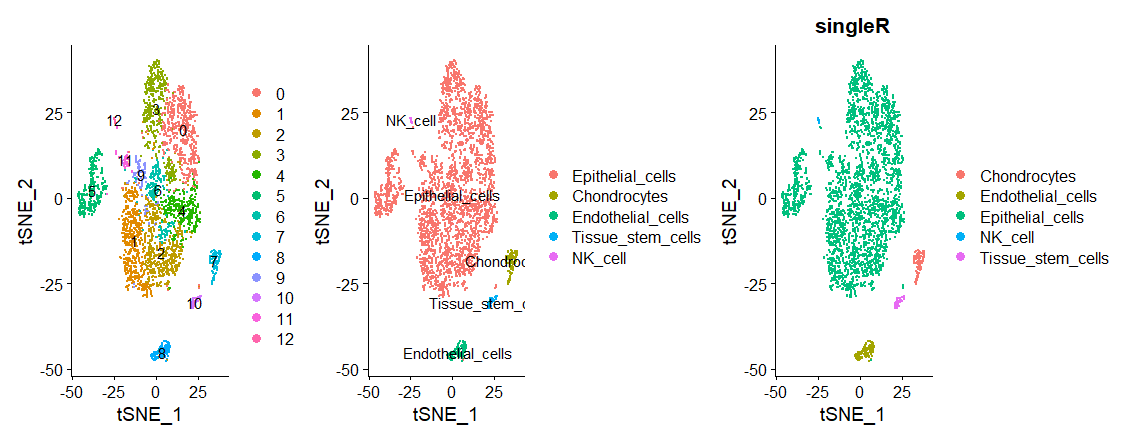
singleR这个包依赖于有较好的细胞注释数据,如果你的样本和它提供的集中差别很多,注释出来还是会有些问题。所以,很多数据,我们需要自己根据marker基因,手动注释,这里后续再介绍。
6.保存数据¶
保存数据,后续教程继续使用。
save(sce1,sce2,file = "data/sce_opt_anno_GSE130001.Rdata")