了解Seurat对象
1.概述¶
单细胞分析世界里数据结构多种多样,主流的四种数据结构分别是Bioconductor主导的SingleCellExperiment,Seurat中的SeuratObject格式,scanpy中的AnnData格式,以及大型数据存储的loom格式。通常一种数据结构对应的内容可以包含所有的分析,例如seurat就可以一用到底,那么我们只要掌握好其中一种数据结构就基本够用。
2.SeuratObject对象¶
有人把seurat对象比作是流水线上的具有不同盒子的容器,经过不同的工序,会在不同的盒子里(slots)增加内容,而这部分工序就叫convertor,包括NormallizeData()、FindVariableFeeature()等,而在每一步工序中对数据进行监管,控制的工具就叫Inspector,包括VlnPlot(),pbmc[['RNA']]@data等。
例如,下面读入的一个seurat对象,那么在这个对象中包含哪些内容呢?

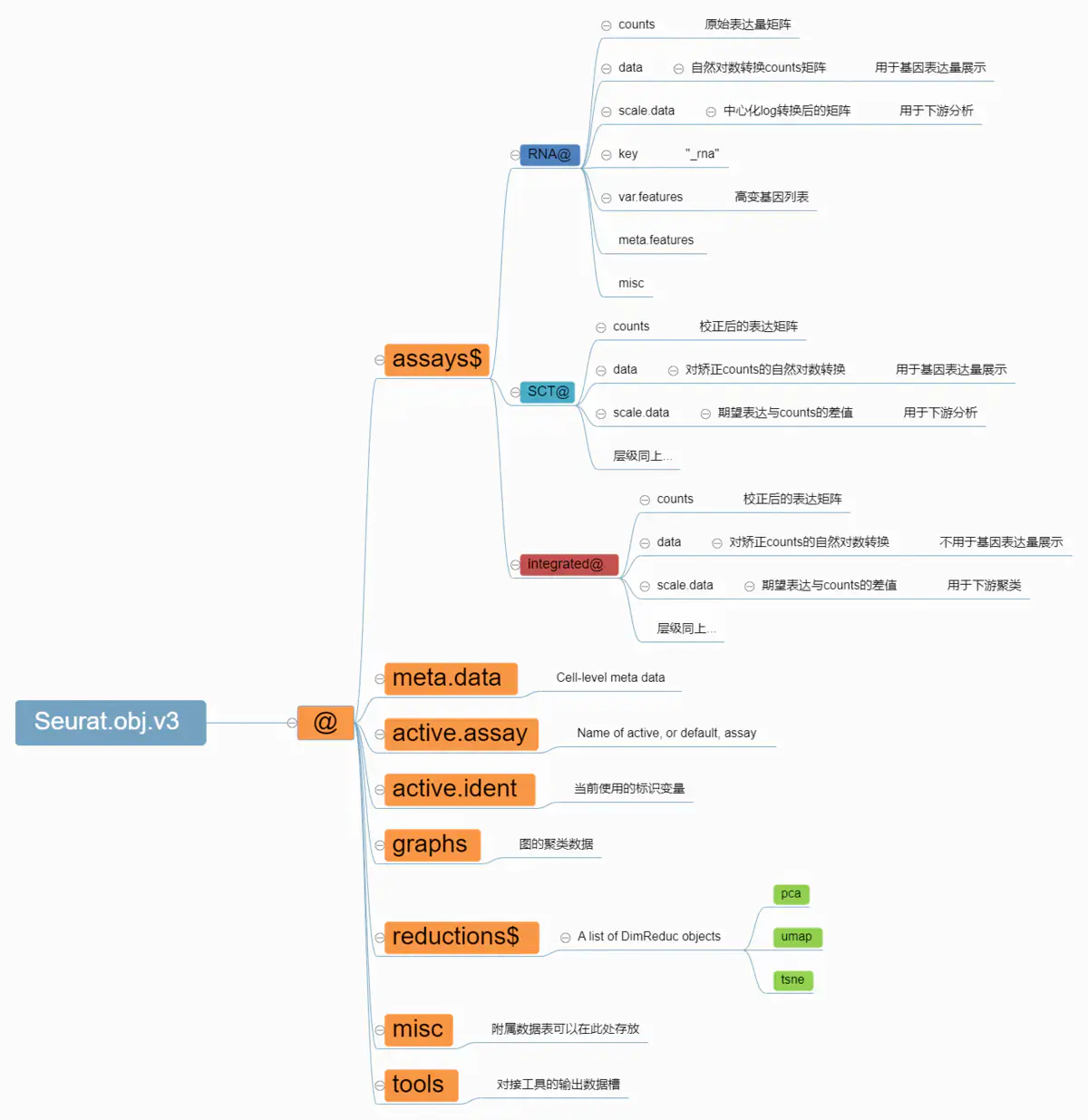
Assays:assays里有一个元素“RNA”,访问assays对象的内部结构,例如pbmc[['RNA']]。
RNA:是assay,其可以包含多个matrix:
Counts:原始的表达量count矩阵; Data:原始数据经过normalized的数据; Scale.data:数据经过scaling后,存放位置; Key:每个assay对象都有一个key,例如‘rna_’; *Var.features:普通向量,高表达变异的基因。
Meta.data:是细胞的注释信息的数据框,行是细胞,列是细胞的属性。 Active.assay:当前激活的assay对象。 标准的seurat流程可以这样进行,包括创建对象,标准化,寻找高突变特征,归一化,聚类等:
# Get cell and feature names, and total numbers
colnames(x = pbmc) ##细胞
Cells(object = pbmc) ##同上获取细胞名称
rownames(x = pbmc) ##获取基因名称
ncol(x = pbmc) ##获取细胞数量
nrow(x = pbmc) ##获取基因数目
可以使用一些函数对行列等进行获取,也可以用Idents获取细胞的分类情况。
# Get cell identity classes
Idents(object = pbmc)
levels(x = pbmc)
table(Idents(pbmc)) ##获取每个细胞类型数目的表格
# Stash cell identity classes
pbmc[["old.ident"]] <- Idents(object = pbmc)
pbmc <- StashIdent(object = pbmc, save.name = "old.ident")
##设置细胞类别
# Set identity classes
Idents(object = pbmc) <- "CD4 T cells"
Idents(object = pbmc, cells = 1:10) <- "CD4 T cells"
# Rename identity classes
pbmc <- RenameIdents(object = pbmc, `CD4 T cells` = "T Helper cells")
另外也可以通过subset来取对象子集,或者通过merge来合并多个seurat对象。
3.Seurat对象中的Assay¶
在-RNA槽: @counts:未作任何处理的原始RNA表达矩阵。
@data:原表达矩阵通过NormalizeData()归一化消除测序文库差异(对于每个细胞,将每个基因的表达量除以该细胞的所有基因表达量之和,然后乘以一个scale.factor, 之后以自然对数进行转换),得到非高斯分布的矩阵。主要用于基因表达量可视化
@scale.data: 通过对log转换后的@data矩阵进行ScaleData()中心化,得到接近于高斯分布的矩阵。
在-SCT槽: @counts:该矩阵是UMI counts矫正后的counts,是由ScaleData(皮尔逊残差)倒推出来的,它是一个回归,运算之后的残差。
@data:矫正后的UMI counts的log-normalized变换,可用于基因表达量可视化。
@scale.data:基因组真实表达量与拟合后的表达预期值的差值(皮尔逊残差:“正则化负二项式回归”的残差)。用于差异表达分析,整合分析,差异基因分析。
SCTransform对测序深度的校正效果要好于log标准化,也可用于矫正线粒体等因素的影响,但不能用于批次矫正。基于概率方法对UMI counts进行建模。在进行了SCTransform操作后,矩阵默认会变成SCT矩阵,如果不加设置,后续的PCA等操作都是基于SCT矩阵。由于SCTransform 更好地估计了方差,并且较高的 PC 维度通常不包括技术变化的影响,所以选择用于聚类的 PC 越多,意味着生物学变化越多。(不同于以往的全局scale归一化方法,需要认真选择用于聚类的PC以避免技术误差建立的PC维度被用于聚类)。
scRNA <- SCTransform(scRNA, vars.to.regress = "percent.mt", verbose = FALSE) #在标准化过程中屏蔽线粒体引起的变异。
簇的Marker基因鉴定最好使用RNA矩阵($RNA@counts)。 sct的到的count并不是真实的基因表达值,而是通过scaledata倒推出来的,它是一个回归,运算之后的残差。
4.归一化和标准化的区别¶
实际上口语里面通常是没办法很便捷的区分这两个概念,我查阅了大家的资料,发现基本上都是混淆在z-score转换和0-1转换这两个上面。
在单细胞 (scRNA-seq) 数据的分析中,进行有效的预处理和标准化非常关键。原始UMI计数不能直接用于比较细胞之间的基因表达,因为它们会被技术和“无意义”的生物变异所混淆。 特别是,观察到的 测序深度 (每个细胞检测到的基因或分子的数量)在细胞之间可能存在显着差异,即使在同一细胞类型内,分子计数的变化可能跨越一个数量级。 虽然现在在scRNA-seq中广泛使用唯一分子标识符 (UMI) 消除了 PCR 扩增偏差,但仍需要通过标准化以消除其他技术变化的影响,如测序深度、细胞裂解和逆转录效率等带来的变异。其实在bulk RNA-seq分析中同样存在,但由于scRNA-seq数据的极度稀疏性,这类问题会更严重。
一般来说,有效归一化分析流程处理后的数据集,基因的标准化表达水平与细胞的总测序深度不相关。下游分析流程(降维、差异表达)也不受测序深度变化的影响。跨细胞归一化基因的方差主要反映生物学异质性,与基因丰度或测序深度无关。例如,标准化后具有高方差的基因应在不同细胞类型之间差异表达,而管家基因应表现出低方差。
所以我这里把归一化和标准化替换成为去除样本/细胞效应或者去除基因效应:
- 首先去除样本/细胞效应:因为不同样本或者细胞的测序数据量不一样,所以同样的一个基因在不同细胞,哪怕你看到的表达量是一样的,但是背后细胞整体测序数据量的差异其实反而说明了这个基因在不同细胞表达量其实是有差异的。在seurat里面的代码是:
# 默认文库大小是1万,默认会取log值。
sce <- NormalizeData(sce, normalization.method = "LogNormalize", scale.factor = 10000)
在使用NormalizeData函数的时候,有一个 normalization.method = "LogNormalize"参数被设置了,这个是为什么呢?
其实很简单,原始的raw counts矩阵是一个离散型的变量,离散程度很高。有的基因表达丰度比较高,counts数为10000,有些低表达的基因counts可能10,甚至在有些样本中为0。因为是单细胞转录组,drop-out现象很严重,其实大量的基因在很多细胞的表达量都是0,如果是10x数据,甚至会出现一个表达矩阵里面97%的数值都是0 的现象。
如果对表达量取一下log10,发现10000变成了4,10变成了1,这样之前离散程度很大的数据就被集中了。
有时当表达量为0时,取log会出现错误,可以log(counts+1)来取log值。当x=1时,所有的log系列函数值都为0。这样原本表达量为0的值,取log后仍为0。
- 然后去除基因效应:这个主要是在绘制热图的时候会需要使用,因为个别基因表达量超级高,在热图里面一枝独秀,实际上我们并不会关心不同基因的表达量高低,我们仅仅是想看指定基因在不同细胞的高低而已,这样的话,就把该基因的表达量在不同细胞的数值,进行z-score这样的转换。这样处理后的表达矩阵,就可以进行后续的降维聚类分群啦。在seurat里面的代码是:
sce <- ScaleData(sce)